Project Overview
Briefly, our project relies on controlling key viral developmental processes in a target-cell specific manner. In our design, the engineered viruses are capable of entering all cells. The viruses are engineered to lack the native copy of a key developmental gene, while containing a second, regulated, copy which is only expressed when the virus infects specific target cells. Thus, viruses infecting non-target cells stall early in their development and are quickly destroyed by the host. Viruses infecting target cells, however, manage to express these essential genes and successfully complete their infection cycle.
As an initial mechanism to target viruses to specific cell types, we will place the viral developmental genes under riboregulator control. Viral mRNAs for the regulated developmental genes will express with a stem loop sequestering ribosome binding sites, preventing translation. Specific mRNA in target E. coli will invade the stem loop, freeing the ribosome binding site and allowing proper translation. We believe this approach is more general than methods which might target specific cell-surface markers. Furthermore, if this method works, it would be possible in principle to extend viral mRNA regulation using aptamers capable of recognizing subtle signals such as post-translational modification.
We selected the viral developmental genes N, Q, and cro as promising targets for regulation. N and Q are antiterminators required for λ to transcribe its full set of genes. Viruses lacking these genes stall at extremely early developmental stages and are completely inviable, barely producing any viral mRNA. cro biases the virus' decision to either lyse a target cell or integrate into the host's DNA, possibly allowing us to selectively 'rewire' host bacteria.
Choosing an appropriate λ strain poses a challenge. Existing strains with defective N, Q, and cro genes lack unique restriction sites to clone our constructs into. Therefore, we will utilize recombineering to introduce introduce these mutations into phages specifically designed to accept cloning inserts.
 An overview of the Caltech 2007 iGEM project. We knock out the native copy of a key developmental gene, then add in a second copy under riboregulated control. Infection will only proceed in cells with the correct trans-activating RNA 'key'.
Project Details
The project consisted of five individual parts, each assigned to one team member, and categorized into one of three independent principles underlying the project:
Current Status
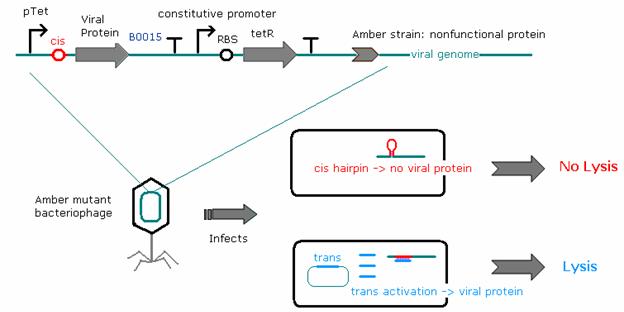
As the recombineering, testing of riboregulators, and titering processes took place concurrently, we needed to find a simpler way to regulate viral protein concentrations in the cells. To this end, E. coli strains have been constructed that contain a low-copy plasmid construct where one of three key developmental viral genes - coding for the cro, N, or Q proteins - is regulated by a tetracycline-dependent promoter. A constitutive promoter produces a steady stream of tetracycline repressor (tetR), which substitutes for the cis repressor in repressing protein levels. The addition of anhydrotetracycline (aTc, acting as the trans activator) inactivates the tetracycline repressor and leads to the production of the respective viral protein in the E. coli cells. This allows us to control the concentration of viral protein produced in the cells by adding varying amounts of aTc to the bacterial growth media.
Titering experiments where cro, N, and Q amber phages were allowed to infect D1210 cells containing the built construct show that heterologous N and Q can complement phages with amber mutations in the respective genes. Adding a cis-repressor to the Q construct lowered production of Q even further, as it eliminated lysis completely. We were unable to express sufficient cro from a plasmid to rescue lytic behavior of the amber cro mutant phage.
Multiple riboregulator designs are being tested (for both activation and repression levels), and successful designs will be cloned into the plasmid constructs. So far, cis construct number 3 and its accompanying trans combinations (cis3/trans1 and cis3/trans2) seem the most promising. Phages resulting from the recombineering process are also being screened for successful N and Q amber mutants.
Future Work
We are currently testing the ability of our best cis/trans combinations (cis3/trans1 and cis3/trans2) to regulate Q expression levels. We will titer the Q amber mutant phage on cells containing the cis3-repressed Q, either with or without the appropriate trans-activating RNA. A positive result (no plaques on cis containing strains, plaques on cis-trans strains) would show successful integration of two independent project components (N/Q protein dependent lysis switch, and the “lock and key” riboregulator). Successful integration demonstrates that standardization on multiple levels (BioBrick parts making each construct, and the more abstract merging of riboregulators into viral decision-making) can allow rapid construction of complex synthetic biological control pathways.
Looking farther forward, we have several tasks to finish. First, we have several more cis/trans combinations to test by flow cytometry. Next, we need to check whether cis repression in the N construct is sufficient to inhibit complementation of N amber mutant phage. As with the cis3-Q/trans1 and cis3-Q/trans2 combinations, we will have to test promising cis/trans combinations with both N and Q.
The final step, then, would be to finish recombineering amber mutations into the N or Q gene of λ-Zap. We will then clone our cis-repressed N and Q constructs into the multiple cloning site of the mutant λ-Zap. These phage will be used to infect E. coli with or without the appropriate trans plasmid. If our riboregulation is sufficient, the phage would only lyse those cells containing the trans plasmid.
Applications
We see several applications for this research. Most immediately, we believe that selective bacteriophage could be used for containment in the event of an accidental environmental release of engineered bacteria. The phage could specifically target the released bacteria, without harming other wild species of bacteria.
Next, the phage could be used in a laboratory setting to help prevent the formation of 'cheaters' in an engineered population. If a genetic construct places host cells at a growth disadvantage, and cells which can eliminate the construct would quickly take over the population. Our phage could be targeted such that they only kill cells which no longer display their desired function, allowing for more direct control over cheating.
Finally, the lessons learned using our system may be applicable to medical applications involving gene therapy. In such an application, we would aim to target specific cells. These could allow tissue-specific treatment, or specific targeting of cancerous cells.
| 
