Experiemental Steps
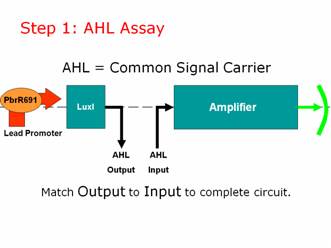
Step 1: AHL Assay
AHL is our common signal carrier between the two main components of our genetic circuit. The output coming from our lead promoter + LuxI is AHL. And the input going into the pLux part of our Amplifier is AHL. To make a working lead detector, the amount of AHL coming out of the first component must be sufficient to turn on the second component. Therefore, as in electrical engineering, we must measure and match our inputs and outputs to create a working genetic circuit.
We used the well characterized part BBa_T9002 for our AHL assay. These cells take in AHL and give us a straight GFP output, without amplification or a positive feedback loop. We created a calibration curve and planned to use these cells to test the amount of AHL coming OUT of our lead promoter in response to different amounts of lead. We would then separately test the amplifier by adding known concentrations of AHL to determine how much AHL is necessary to give us a GFP output. Then we can put both pieces of information together to give us a lead sensor.\
Registry Links:
[http://partsregistry.org/Part:BBa_T9002 BBa_T9002]
[http://partsregistry.org/Part:BBa_F2620 BBa_F2620]
Step 2: Characterize Amplifier
We characterized our amplifier and found some very unexpected results!
The main difference between J37015 and T9002 is that there is a positive feedback loop in J37015. So we would expect J37015 to make much more GFP than T9002 at the same AHL concentrations. However, J37015 actually made LESS GFP.
Also, in T9002, we noticed that adding more AHL gave us more GFP, as was to be expected. However in J37015, we found that adding more AHL gave us LESS GFP.
Clearly, the amplifier did not work the way we had thought it would. This highlights the importance of part characterization. If synthetic biology is going to advance, parts in the registry must be well characterized to be useful for engineering purposes. We took it upon ourselves to characterize this part to ensure that others would not run into the same problems we did.
[http://partsregistry.org/Part:BBa_J37015:Experience See our extensive characterization of this part, J37015, on the Registry for the various experiments we conducted.]
Step 3: Promoter and Lead Binding Protein
Despite our amplifier results, we were not deterred. We still hoped to create a lead promoter and lead binding protein BioBrick to make our original simple lead detector.
We obtained the genome of Ralstonia metallidurans. We then used PCR to pull out 15 different variations of the lead binding protein and lead gene.
The diagram shows what the top and bottom strands of the genome where the lead binding protein, PbrR691 and the lead promoter (within this Non-Coding Region) lie. PbrA is a resistance gene that was not involved in our system but is relevant to the genome.
According to the literature, the forward lead promoter is within this noncoding region. After communicating with the group that originally found this protein, we also suspected that a constitutive promoter region for PbrR691 could be found in the reverse direction in this noncoding region. Therefore, we used PCR and cloning to simply combine the PbrR691 coding region and blue noncoding region to GFP, hoping that we would not need to express PbrR691 under pTet’s constitutive control to get a responsive lead detector.
We also used PCR to obtain separate copies of the PbrR691 coding region and the lead promoter.
In the end we had 15 different variations of parts that we could add to the registry:
3 = just the Lead Binding Protein, PbrR691.
3 = just the Lead Promoter
1 = the Lead Promoter + some small amount of PbrA with a stop codon. (We included about 100 bp of PbrA because we were afraid that pulling out the promoter alone might disrupt the distance between the protein start codon and the RBS within the non-coding region, which must be very specific for expression of the protein)
6 = Lead Binding Protein + Promoter (non-coding region), as per the method suggested to us by the original research group
2 = Lead Binding Protein + Promoter + 100 bp of PbrA
Plans after PCR
Planned Ligations
- All 15 parts into BioBrick plasmid
- pTet to PbrR691 alone
- All promoters and PbrR691 combinations to LuxI
- All promoters and PbrR691 combinations to GFP
- pTet-PbrR691 to promoters-LuxI
- pTet-PbrR691 to promoters-GFP
Completed Ligations
- All 15 parts into BioBrick plasmid
- All promoters and PbrR691 combinations to LuxI
- All promoters and PbrR691 combinations to GFP
These ligations must be tested to see if the parts actually work.
Our Preliminary Results
PbrR691/promoter combinations in presence of lead nitrate give no GFP production and no AHL production compared to control. Therefore, we decided to abandon that method of creating a lead detector.
Future Plans:
- Finish ligating Lead Binding Protein to pTet, where it can be constitutively expressed.
- Finish ligating Lead promoter to GFP.
- Put both of the above components onto a single plasmid, transform E coli to generate a simple lead detector!
|





