From 2007.igem.org
Mississippi State/Project Description
For more information relating to Mississippi State iGEM 2007 and other synthetic biology projects, please visit the link below!
- [http://www.abe.msstate.edu/wiki/msusbcwiki/index.php/Main_Page Mississippi_State/Mississippi State University Synthetic Biology Club]
Mississippi State University Synthetic Biology Club (MSUSBC)
The Mississippi State University Synthetic Biology Club (MSUSBC) was formed on January 18, 2007.
Its initial members consist of students and faculty from Biological Engineering, Biochemistry, and Microbiology.
The club had its first meeting on January 18, 2007, in the Biological Engineering Building.
|

Welcome to [http://www.msstate.edu Mississippi State University!]
|

Standing (L to R): Scott Tran, Sam Pote, Victor Ho, Robert Morris, Dr. Din-Pow Ma, Joe Chen, James Kastrantas, and Dr. Filip To. Kneeling: Karen Parks and Lauren Beatty.
|
The Team
Faculty Members:
Dr. Filip To Agricultural and Biological Engineering
Dr. Tod French Chemical Engineering
Dr. Din-Pow Ma Biochemistry
Undergraduate Students:
Lauren Beatty Senior, Biomedical Engineering
Scott Tran Senior, Biological Engineering
Joe Chen Senior, Biological Engineering
James Kastrantas Senior, Biochemistry
Karen Parks Senior, Biological Engineering
Caleb Dulaney Junior, Biological Engineering
Sam Pote Sophomore, Biological Engineering
Graduate Students:
Robert Morris Masters student, Biological Engineering
Meng-Hsuan Ho Doctoral student, Molecular Biology
[http://www.abe.msstate.edu/index.php Biological Engineering Department]
[http://www.msstate.edu/dept/biochemistry/ Biochemistry Department]
|
Introduction
- International Genetically Engineered Machine (iGEM) is a student-led competition to build the most innovative "machine" by synthetic biology.
- Headquarters is located at the Massachusetts Institute of Technology.
- In 2006, 37 schools and over 400 students from around the world participated in projects to construct biologically engineered systems.
- Task of each team is to apply engineering methodology to design and develop a new biological system ("machine") through the use of existing and/or newly formed microscopic biological parts (termed BioBricks).
- Type of the "machine" is chosen by each individual school participating, and the only criterion is that the "machine" be made entirely of the functional units of DNA called BioBricks.
- A registry of all BioBricks is kept in the MIT Registry of Standard Biological Parts, which is regularly updated to include new parts developed by teams.
- Parts for each iGEM team are obtained through the Registry for a fee.
- Annual Jamboree for students to present their projects takes place at MIT in November.
iGEM 2006: Genetically Engineered H2 Detector
[http://parts2.mit.edu/wiki/index.php/Mississippi_State_University_2006
View Last Year's Project]
|
iGEM 2007: Ubiquitin-Ligase Probe
Fluorescent Assay of RING-Type Ubiquitin-Ligase
Overview
- The search for renewable energy sources from alternative crops has led to the desire to understand the pathway by which lipids are synthesized. Higher lipid content has a direct correlation to higher energy value, so if the plant pathways involved in lipid metabolism can be modulated, biofuels could become more economically feasible. It has been reported that the ubiquitin proteasome pathway affects lipid metabolic processes. The ubiqination of target proteins for degradation requires the sequential activity of three enzymes: a ubiquitin activating enzyme (E1), a ubiquitin conjugating enzyme (E2), and a ubiquitin ligase (E3). The objective of the 2007 MSU iGEM project is to develop a rapid and sensitive method for assaying the ubiquitin ligase (E3) activity.
- The E3 activity is generally determined by using an in vitro ubiquitination assay. The E3 is first incubated with ATP, ubiquitin, and wheat germ lysate containing E1 and E2 activities (or purified E1 and E2). The ligase reactions are then fractionated by electrophoresis on an SDS-PAGE gel and subjected to Western blotting for the detection of high molecular weight polyubiquitinated E3 proteins. This detection method is costly and time consuming.
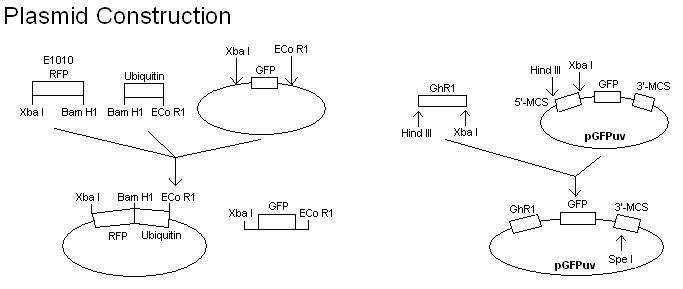
- To develop a quicker assay of E3 activity, the E3 coding region is cloned into a plasmid and expressed as an in-frame C- (or N-) terminal fusion with a GFP protein. The E3 activity is then assayed by directly irradiating the protein gel with long wave ultraviolet light. The expression of green fluorescence in multiple, high molecular weight protein bands would indicate the ligase activity. To confirm that the multiple protein bands are ubiquinated, a plasmid containing RFP in frame with ubiquitin will also be constructed and transformed into E. coli cells harboring the E3-GFP fusion.
- Future work will involve in designing universal plasmids to provide for a quicker assay of any E3 ligases.
|
Project Goals
- Fuse GFPuv to GhR1 (the gene corresponding with our E3 of choice).
- Clone GhR1-GFPuv gene into plasmid.
- Sequence to check.
- Fuse RFP to ubiquitin.
- Clone RFP-ubiquitin gene into separate plasmid.
- Sequence to check.
- Express proteins in the cell. They will interact with each other and the poly-ubiquitin chain will grow.
- Lyse the cells.
- Add components for binding.
- Run gel.
- Analyze gel under UV light.
- If the protein band is tagged with GFPuv, it will be visible. The expected outcome of expression of both GFPuv and RFP is amber colored light.
Alternatives to co-transforming 2 plasmids into the cell:
a) Perform 2 transformations in 1 pET plasmid.
b) Find another plasmid that has a location to clone RFP into.
Problems Encountered + Solutions
- June 22 - Ubiquitin-J01095 Construction
- July 6 - Low GhR1 Expression in Protein Gel
- July 23 - Unsuccessful Amplification of Ubiquitin
- July 25 - Lack of Protein Separation
- July 31 - Incomplete Protein Expression
- August 8 - Restriction Site Within GhR1 Gene
- August 28 - Incorrect GhR1 Sequence
|
Accomplishments
- September 20 - Repeated subculture.
- September 19 - Subcultured cells and induced with IPTG.
- September 18 - Overnight culture of BL21(DE3).
- September 8 - Picked 4 colonies from previous plates, 2 GhR1-S and 2 GhR1-F.
- September 3 - Subcultured cells. Native protein gel (8% Separating) using GFPuv as the control.
- September 2 - Analyzed sequence. Overnight culture of BL21(DE3).
- September 1 - Thermal cycling and sequencing of GhR1-S and GhR1-F.
- August 31 - Extracted plasmids from BL21(DE3) cells. Confirmed by agarose gel and optical density testing.
- August 30 - Picked colonies off plates, 3 GhR1-S and 3 GhR1-F.
- August 28 - Prepared competent cells of BL21(DE3). Purified gel extraction of digests of pGFPuv, GhR1-S, and GhR1-F. Ligation of GhR1-S and GhR1-F into pGFPuv. Transformed cells were grown overnight.
- August 26 - Sequencing of GhR1-S and GhR1-F constructions to confirm chimera sequence.
- August 25 - Isolation of GhR1-S and GhR1-F constructions from XL1-Blue. Agarose gel ran to confirm sizes.
- August 19 - Extracted plasmids from XL1 Blue cells. Confirmed by agarose gel.
- August 18 - Cultivated and inoculated colonies.
- August 17 - Prepared competent cells. Purified gel extraction of digests of pGFPuv, GhR1-S, and GhR1-F. Ligation of GhR1-S and GhR1-F into pGFPuv.
- August 16 - Purified gel extractions of 4 samples. Confirmed by agarose gel. Double digestion of GhR1-S and GhR1-F into pGFPuv. Ran agarose gel. Extracted from gel.
- August 15 - PCR on 4 samples: GhR1-S(short), GhR1-F(full), ubiquitin-Taq, and ubiquitin-PFU. Ran two agarose gels of PCR products. Extracted from gel.
- August 14 - Reverse transcription PCR on ubiquitin from cDNA. PCR on ubiquitin (using both Taq and PFU). Extracted and purified ubiquitin from gel.
- August 13 - PCR on ubiquitin from cDNA using Taq polymerase. Extracted from gel.
- August 8 - Purified gel extraction. Digested both sizes of GhR1, along with pGFPuv.
- August 7 - Confirmed on gel. Performed thermal cycling of ubiquitin. PCR on GhR1 (both sizes). Extracted from gel.
- August 6 - Confirmed extraction and purification of ubiquitin. PCR on ubiquitin. Extracted and purified from gel.
- August 3 - PCR on ubiquitin from cDNA using PFU polymerase. Extracted and purified ubiquitin from gel.
- August 2 - Reverse transcription PCR on ubiquitin from resultant cDNA.
- August 1 - Ran agarose gel of RNA to confirm its quality.
- July 31- Confirmed extraction and purification of ubiquitin. PCR on ubiquitin.
- July 30- Ran agarose gel of ubiquitin. Extracted ubiquitin from gel.
- July 27- Confirmed extraction and purification of E1010 and pGFP. Repeated western blot procedure from July 25.
- July 26- Digested E1010 and pGFPuv. Extracted them from gel. Ubiquitinated GhR1-GFPuv fusion on native protein gel (8% Separating) using GFPuv as the control.
- July 25- Performed western blot on native protein gel containing GhR1-GFPuv and GFPuv both with and without ubiquitination.
- July 24- Ubiquitinated GhR1-GFPuv fusion on native protein gel (10% Separating) using GFPuv as the control.
- July 23- Extracted and purified E1010. Confirmed by running gel. Extracted ubiquitin from gel.
- July 20- PCR on E1010 and ubiquitin. Native protein gel of GhR1-GFPuv fusion in BL21(DE3).
- July 19- Extracted ubiquitin from gel.
- July 18- Native protein gel electrophoresis.
- July 17- Purified PCR mixture. Repeated transformation of E1010 into XL1-Blue.
- July 16- Confirmed extraction of BL21(DE3) with GhR1-pGFPuv. PCR on GhR1-pGFPuv.
- July 11- Transformed GhR1-pGFPuv into BL21(DE3) and E1010 into XL1-Blue.
- July 10- Subcultured cells overnight. Ordered primers for E1010 (RFP) and ubiquitin.
- July 9 - Analyzed protein interaction test. Inoculated cells of the strain BL21(DE3). New construction defined.
- July 6 - Tested protein interaction by native protein gel electrophoresis.
- July 5 - Analyzed protein expression test.
- July 3 - Tested protein expression of GhR1-GFPuv using GFPuv as the control.
- July 2 - Inoculated cells containing GhR1-pGFPuv.
- June 22- Confirmed sequence of GhR1-pGFPuv.
- June 19- Sequenced J01095. PCR on GhR1-pGFPuv.
- June 18- Confirmed GhR1-pGFPuv ligation. Thermal cycling of J01095.
- June 15- Extracted GhR1-pGFPuv plasmid.
- June 14- Inoculated E.coli + plasmid into liquid media.
- June 13- Extracted GhR1 and pGFPuv from gel. Ligated and transformed GhR1-pGFPuv.
- June 12- Digested GhR1 and pGFPuv.
- June 11- Extracted GhR1 from gel.
- June 5 - PCR on GhR1.
- May 30 - Extracted plasmids J01095 and J04500.
- May 29 - Harvested cells and performed electrophoresis.
- May 28 - Cultivated and inoculated colonies.
- May 24 - Transformed DNA into E. coli (XL1 Blue strain).
- May 23 - Began lab work. Removed chosen parts from DNA plates.
- May 22 - Student meeting, BioBrick parts for assembly identified.
- May 21 - Specific project goals defined.
- May 14 - Meeting with faculty to discuss parameters and define project.
|


