Recombineering
Background
Recombineering (recombination-mediated genetic engineering) is a recently developed in vivo technique for making recombinant DNA. As a fast and efficient alternative to classically used in vitro techniques, recombineering takes advantage of lambda phage's homologous recombination proteins collectively known as Red. Previous genetically engineered systems could not successfully insert linear DNA into E. coli due to degradation by nucleases. However, homologous recombination of ssDNA succeeded in the presence of the Red proteins, which inhibited the degrading nuclease in E. coli. Therefore, a defective lambda prophage was engineered with lysis and replication functions inhibited and Red functions retained. Cells containing this prophage facilitate homologous-recombination based engineering of novel genetic sequences, hence the term recombineering.
One can adapt recombineering to introduce short mutations into lambda phage genomes[1]. Briefly, E. coli cells expressing the proper prophage gene suite are infected with the lambda phage strain to be engineered. The cells are then made electrocompetent and single stranded oligos (about 70 nt) containing the desired mutation, plus flanking homology regions, are electroporated in. The new phages, a mixed population of wild type and engineered, are then allowed to lyse the host cells. The resulting lysate is screened in an appropriate manner to detect the mutation of interest.
In our particular application, we are introducing amber stop codons in frame to N and Q. To screen for these mutations, phages are plated for plaques in an amber suppressor strain (E. coli LE392, supF). A further layer of non-amber suppressing cells (E. coli MG1655), without phages, is immediately plated on top of the amber suppressing layer. Phages without the amber mutation will make plaques that 'punch through' both layers, while amber mutant phages will be unable to penetrate the upper layer. The two types of plaques can be distinguished by eye, allowing us to screen for amber mutants.
Integration
We will use recombineering to create phages which will serve as suitable background strains for this project.
Our project requires phage strains with two main characteristics. First, our phage strains must be defective in expression of the N, Q, or cro developmental genes, while still being easy to propagate. Second, the strains must allow easy cloning of heterologous constructs -- our riboregulated N, Q, and cro expression constructs -- into them.
Recombineering allows us to satisfy the first constraint in an elegant way. Specifically, we will use recombineering to insert in-frame amber stop mutations into the coding sequences of N, & Q or cro. The amber stop mutation prevents successful translation of these genes in most E. coli strains, crippling the phages. By stopping translation with only a single point mutation, we minimize the mutation's impact on other functions in the densely coded phage genome[2]. Finally, phages with amber stop mutations can be easily propagated in special amber suppressor E. coli strains, a standard decades-old technique in classical lambda genetics.
To satisfy the second constraint, we choose a phage strain commonly used for cloning cDNA libraries. This phage strain, named Lambda Zap (by Stratagene)[3] is engineered to contain unique restriction sites in nonessential areas of the phage genome. This makes the strain much easier to work with than standard lambda strains, which contain few uniques restriction sites, often in the middle of critical genes. Note that we chose to work with a commercial strain for convenience, and that our method can be easily ported to freely available lambda phage cloning strains.
Status and Future Plans
We designed oligos to introduce appropriate point mutations into N, Q, and cro. As recombineering efficiency has been estimate to be on the order of 1%[1], we put considerable effort into optimizing our screen. Initial tests, against a mixed population of 50% cI60 mutants (obligate lytic) and 50% N or Q amber mutants, revealed that the standard double-layer titering assay showed only subtle differences between the two plaque types. "Cloudy" and "clear" plaques provded somewhat difficult to distinguish by eye.
We therefore attempted a slight modification, labeling the non-suppressing bacterial with a high copy plasmid driving an RFP. This process did indeed make the two plaque types more distinguishable with the aid of a fluorescence dissecting scope. Microscopy results confirm that the two plaque types do indeed correspond to different degrees of lysis in the two layers. Below on the left is an obligate lytic mutant (cI60), which creates plaques in both unlabeled suppressor (white) and RFP labeled non-suppressor cells (red). Below on the right is an N-amber mutant phage creating a plaque in the unlabeled suppressor cells, while leaving the RFP labeled non-suppressor cells intact.
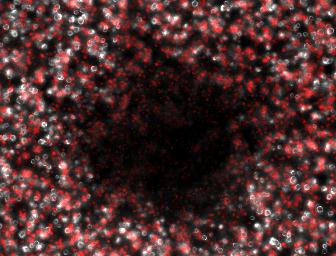 Clear Plaque Clear Plaque Cloudy Plaque Cloudy Plaque
Relevant Protocols
References
- Oppenheim AB, Rattray AJ, Bubunenko M, Thomason LC, and Court DL. In vivo recombineering of bacteriophage lambda by PCR fragments and single-strand oligonucleotides. Virology 2004 Feb 20; 319(2) 185-9. doi:10.1016/j.virol.2003.11.007 pmid:14980479. PubMed HubMed [oppenheim]
- Chan LY, Kosuri S, and Endy D. Refactoring bacteriophage T7. Mol Syst Biol 2005; 1 2005.0018. doi:10.1038/msb4100025 pmid:16729053. PubMed HubMed [chan]
- Short JM, Fernandez JM, Sorge JA, and Huse WD. Lambda ZAP: a bacteriophage lambda expression vector with in vivo excision properties. Nucleic Acids Res 1988 Aug 11; 16(15) 7583-600. pmid:2970625. PubMed HubMed [short]
|




