Ljubljana/finalsystem
From 2007.igem.org
Performance of the Final Functional Systems
Split systems based on heterodimerization
After all subsystems were tested to be functional and their expression was optimized, final tests were performed to demonstrate that split-ubiquitin and split TEV protease systems were active. For the system to work there are some crucial steps:
Our results demonstrate that split-ubiquitin and split TEVP systems were activated by the HIV coat protein gp120 which causes dimerization of CD4 and CCR5 receptors. Activation was amplified by the T7 RNAP system connected to luciferase reporter. Figures 4 and 5 show normalized luciferase activities of gp120-stimulated cells, nonstimulated and control cells, respectively in the split ubiquitin (Fig. 4) and split TEVP (Fig.5) system.

Fig. 4. Viral envelope protein gp120 activates the split ubiquitin system. Cells were transfected with CMV–CD4–CUb–T7RNA polymerase, CMV–CCR5–Nub, pT7-fLuc and CMV-rLuc reporter plasmids. 12 hours after transfection cells were activated with gp120 protein for additional 12 hours. Cells were lysed and luciferase activity was measured. The results were normalized for transfection efficiency and cell number using rLuc readings. Addition of gp120 clearly activated the split ubiquitin system as higher amounts of T7 promoter-regulated luciferase activity were measured. Luminescence was observed also in cells which were not stimulated, probably because of small level of spontaneusly dimerized CD4 and CCR5, as we used relatively strong promoter, which may cause high level receptors expression at the plasma membrane. T-test: p<0,01, ***; p<0,005, ****
.

Fig. 5. Viral gp120 activates the split TEV protease system. Cells were transfected with CMV–CD4–TEVC, CMV–CCR5–TEVN, myristoylation signal-TEVprotease site–T7 protease, pT7-fLuc and CMV-rLuc reporter plasmids. 12 hours after transfection cells were activated with gp120 protein for additional 12 hours. Cells were lysed and luciferase activity was measured. The results were normalized for transfection efficiency and cell number using rLuc readings. Addition of gp120 activated split TEV protease system as higher amounts of T7promoter luciferase were measured. Increased luminiscence was also observed in control cells, probably due to the same reason as in split ubiquitin experiment. T-test: p<0,1, *.T-test: p<0,005, ****.
Interpretation of results:
Firefly luciferase gene is regulated by the inducible T7 promoter, while renilla luciferase is under constitutive CMV promoter. The difference in luminescence of both luciferases is a consequence of different transfection efficiencies and cell number in different wells. Normalization means that we divide relative luminescence of firefly luciferase by relative luminescence of renilla luciferase. The ratio we get presents differences in promoter activity – the higher the ratio the more active promoter we have.
Measurements of luminescence revealed a higher firefly/renilla ratio in cells stimulated with viral protein gp120. A significant difference between stimulated and nonstimulated cells was observed. This is a strong evidence that the engineered device actually works as planned.
As seen in Figure 1, we could prove enhanced activation of T7 promoter in gp120 stimulated cells. To verify that the response is due only to T7 RNAP liberated from the ubiquitin fusion (and not e.g. by promoter leaking, spontaneous dimerization of CD4/CCR5, unspecific cleavage of membrane-attached T7 polymerase or unexpected activity of membrane-anchored T7 RNAP, we performed two additional control experiments.
Control 1: Cells were transfected with T7 promoter – firefly luciferase vector (and renilla luciferase for normalization standard). Only little background luminescence was detected. Control 2: Cells were transfected with both luciferase constructs and with CD4-CUb-T7 RNAP (control column in Figure 1). Luminescence was low, meaning that neither leaking of T7 promoter nor activity of T7 RNA polymerase on the plasma membrane (or its liberation from the bilayer) are the cause of T7 promoter activation.
Luminescence was observed also in non-stimulated cells. This could be due to spontaneous dimerization of CD4 and CCR5, probably because we used a relatively strong promoter which could lead to too many receptor molecules expressed and bound to the plasma membrane.
In the comparable experiment just with the split-TEVP attached to CD4 and CCR5 and T7 polymerase attached to the plasma membrane via a myristoyl group and connected to the receptor-split TEVP by a TEVP cleavage site. Results, presented in Figure 2 are highly comparable to those obtained with split ubiquitin, were controlled and interpreted as presented above for the split ubiquitin system.
In conclusion, with these experiments we proved that stimulation by HIV viral coat protein activated cellular response through both split-protein devices. We can expect that by replacing luciferase by an effector gene (caspase 3 or ß-interferon) cells would respond by activation of apoptosis or by synthesis of antiviral substances which would prevent the virus from spreading infection to healthy cells.
Fluorescent confocal microscopy was used to demonstrate the performance of the split TEV protease system and confirm luciferase data. Cells were transfected with CMV–CD4–TEVC, CMV–CCR5–TEVN and myristoylation signal–TEVprotease site–mCerulean. Fluorescence was detected at the plasma membrane, as expected (Fig. 6C). 22 hours after the transfection cells were stimulated with pseudovirus or gp120 protein and again observed 18 hours later. mCerulean was released from the membrane into cytosol in both cases (Fig. 6A and 6B). Additionally, membrane was stained after the stimulation to confirm the cytosolic localization, respectively (Fig. 6B).

Fig. 6. HIV pseudovirus and gp120 protein activate split TEV protease system. HEK293T cells were transfected with CMV–CD4–TEVC, CMV–CCR5–TEVN, myristoylation signal–TEVprotease site–mCerulean. 22 hours after transfection cells were stimulated with pseudovirus (A) or gp120 protein (B) for additional 18 hours. In both cases the split TEV protease system was activated and mCerulean reporter protein was released from membrane to cytosol. However, in unstimulated cells the system remained inactive and mCerulean was detected mainly at the plasma membrane (C).
We proposed that analogously T7 RNA polymerase fused to the receptor instead of mCerulean will be cleaved. Transfection with TEV protease cleavage site fused with T7 RNA polymerase with NLS and T7promoter–mCerulean was performed. Cells were stimulated with pseudovirus or gp120 protein 22 hours after transfection and again observed 18 hours later (Fig. 7). In stimulated cells intense expression of mCerulean was detected (Fig. 8A and 8B)whereas unstimulated cells remained clear (Fig. 7C). The system, using receptors CD4 and CCR5 with truncated cytoplasmic domains was also tested and resulted in similar results.
These experiments confirm that when activated, split TEV protease system releases T7 RNA polymerase from the membrane. Consequently T7 RNA polymerase translocates to the nucleus where it transcribes genes under the T7 promoter.

Fig. 7. Activated split TEV protease system transcribes genes under the T7 promoter. HEK293T cells were transfected with CMV–CD4–TEVC, CMV–CCR5–TEVN, myristoylation signal–TEVprotease site–T7 RNA polymerase-NLS and reporter plasmid T7-mCerulean. 22 hours after the transfection cells were stimulated with pseudovirus (A) or gp120 protein (B) for additional 18 hours. In both cases the split TEV protease system was activated, resulting in translocation of T7 RNA polymerase from the plasma membrane to the nucleus. Consequently mCerulean reporter protein was transcribed and detected in cytosol. In unstimulated cells the system remained inactive and no fluorescence at all was observed (C).
System based on the activation by HIV protease
The process of system activation was observed using fluorescent confocal microscopy. Cells were transfected with CMV–CD4–HIV protease cleavage site–mCherry–NLS and optionally cotransfected with HIV protease on pNL4.3 plasmid. In cells without HIV protease fluorescence was detected at the plasma membrane (Fig. 8A), whereas in HIV protease cotransfected cells, fluorescent protein mCherry was released from the membrane and translocated to the nucleus (Fig. 8B). Similar results were obtained when mCherry was substituted with T7 RNA polymerase and additional construct pT7–mCerulean was transfected to detect the T7 RNA polymerase activity. In cells with HIV protease cotransfection, mCerulean was detected in cytosol (Fig. 9B), but no fluorescence at all was observed in the control experiment without of the HIV protease activity (Fig. 9A).
The system, using myristoylation signal instead of CD4 to anchor the T7 RNA polymerase to the membrane was also tested and we obtained similar results (data not shown).
We can conclude that in the presence of HIV protease, T7 RNA polymerase is released from the membrane and translocates to the nuceleus where it transcribes genes under the T7 promoter.
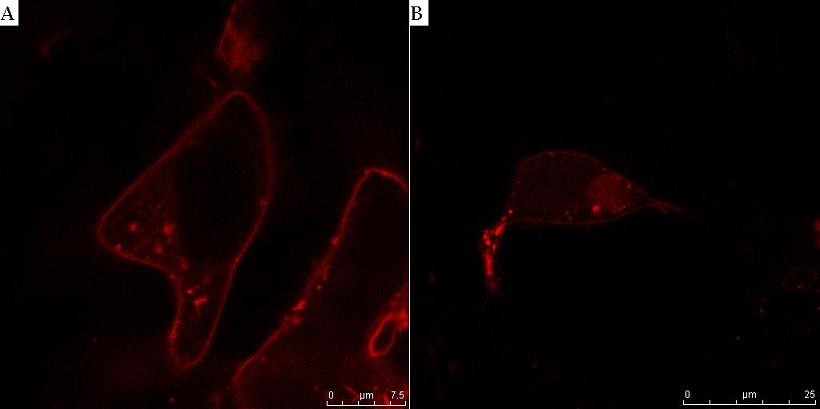
Fig. 8. HIV protease cleaves the linker between the membrane anchor and mCherry. HEK293T cells were transfected with CMV-CD4-HIVprotease cleavage site-mCherry-NLS (A) and additionally with HIV protease plasmid pNL4.3 (B). In cells without HIV protease, membrane localization of mCherry reporter protein was observed (A), but in the presence of HIV protease mCherry-NLS was released to cytosol and translocated to nucleus (B).

Fig. 9. T7 RNA polymerase is activated by the cleavage of the linker on the membrane anchor by HIV protease. HEK293T cells were transfected with CMV-CD4-HIVprotease cleavage site-T7 RNA polymerase-NLS, reporter plasmid T7-mCerulean (A) and additionally with HIV protease plasmid pNL4.3 (B). HIV protease cut off T7 RNA polymerase from the membrane, thus activating it. Consequently mCerulean under the T7 promoter was transcribed and detected in cytosol (B). In the absence of protease the system remained inactive and no fluorescence was observed (A).
Effector protein – caspase-3
Instead of reporter proteins effector proteins such as caspase-3 were used, which are close to the real application. Caspase-3, a cysteine peptidase, which is the main executioner of the apoptosis. We have selected caspase-3 since we wanted to induce apoptosis in HIV-infected cells so that HIV could not replicate and infect the surrounding cells. Apoptosis prevents any inflammation to the neighboring cells and provides for a "clean removal" of apoptotic cells.
Two constructs were prepared for testing the caspase-3 activity: CMV-Cas3 and pT7-Cas3. The first construct was used to test if the caspase-3 is active in transfected cells. The second construct with caspase-3 under the T7 promoter was used as our main effector. Caspase-3 will be transcribed only when active T7 RNA polymerase is present in cytosol or nucleus. This should happen when our devices are activated by HIV virus-induced receptor heterodimerization or by HIV protease, expressed in HIV infected cells.
ELISA was used to determine the amount of caspase-3 in cells – this indicates the apoptotic potential of our system. We wanted to show that amount of caspase-3 in cells infected with HIV protease is greater than in non-infected cells (Fig. 10). The amount of caspase-3 had to be carefully monitored since the expression of caspase-3 leads to apoptosis and killing the cells, which we then can not detect anymore. So, timing of ELISA tests was therefore very important.

Fig. 10. Caspase-3 expression is enchanced in HIV-protease transfected cells. HeLa cells were transfected with effector gene pT7-Cas3 alone or with effector gene and components of system based on activation by HIV protease activity: CD4 membrane receptor with bound T7 RNA polymerase via HIV protease cleavage site and HIV protease in pNL4.3 vector. ELISA test was made with antibodies against human caspase-3. Significant amount of caspase-3 was detected in nontransfected cells and in cells, transfected with pT7-Cas3 as well. Since ELISA can also detect inactive and endogenous caspase-3 and since certain amount of cells is always in apoptosis, we did not expect extremely low values in these two samples. Significantly elevated levels of caspase-3 were detected in cells, transfected with our system components, meaning that more caspase-3 is transcribed and activated cells after HIV protease cuts off T7 RNA polymerase from transmembrane receptor. In terms of our initial idea this means that HIV entry and HIV protease activity in infected cells could trigger apoptosis and thus prevent replication of virus. T-test: p<0,05, **; p<0,01, ***.
Flow cytometry using annexin V staining was used for detection of amount of apoptotic cells as a consequence of caspase 3 activity after system activation. We have transfected cells with components of our systems (that have been previously proved as functional) with caspase-3 under T7 promoter as an effector gene. As a positive control we used cells, transfected with CMV-Cas3 construct. Activation of apoptosis varies between different cell lines and also the efficiency between different inducers varies significantly. Although some apoptosis was detected still an additional optimization is needed to obtain reliable results.
Instead of caspase-3, which triggers apoptosis in HIV infected cells we could use effector proteins that would act as antiviral agents. Good example is interferon-β. Interferons are very important in antiviral defense. They are usually secreted as a consequence of elevated levels of dsRNA in cells. Its effects include prevention of viral replication and stimulation of the immune response. In extreme cases it can also trigger apoptosis, probably trough increased production of p53 gene product.
We have not directly tested the activity of human interferon-β in HIV-infected cells but have deposited BioBricks containing interferon-β, pT7-interferon-β and CMV-interferon-β to the Registry.